Disordini genetici del sistema HDL
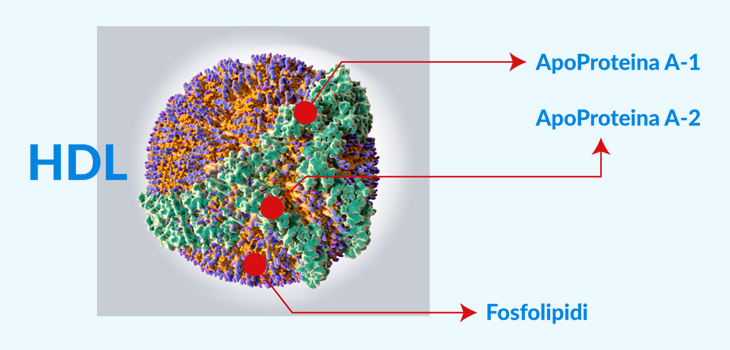
Oggi parliamo un po’ di genetica! Abbiamo conosciuto meglio e più da vicino le tre funzioni fondamentali delle lipoproteine HDL:
- Trasporto inverso del colesterolo
- Protezione endoteliale
- Attività antiossidante
Cosa accade in caso di mutazioni che alterano delle proteine del sistema HDL?
I disordini genetici del sistema HDL sono rare patologie monogeniche rare causate da mutazioni in geni che codificano per diverse proteine coinvolte nel metabolismo delle HDL.
I disordini genetici di HDL non sono solo caratterizzati da livelli di HDL circolanti molto ridotti o molto elevati, ma anche da alterazioni di vario tipo delle sottoclassi di HDL
Le più comuni mutazioni genetiche note coinvolgono i seguenti geni:
- i geni ABCA1, APOA1 e LCAT, che causano ipoalfalipoproteinemia
- il gene della CETP, che causa iperalfalipoproteinemia.
Mutazioni del gene ABCA1
Il gene ABCA1 è localizzato sul cromosoma 9 (9q31) e codifica per una proteina di membrana di 2.261 residui aminoacidici appartenente alla famiglia dei trasportatori ABC. Ad oggi, in letteratura sono state riportate più di 170 diverse mutazioni del gene ABCA1 in grado di modificare in maniera differente la funzionalità della proteina ABCA1. Mutazioni a carico di entrambi gli alleli del gene ABCA1 (omozigosi) causano la malattia di Tangier, una rara patologia di tipo recessivo caratterizzata da un’assenza quasi completa di HDL. Uno degli aspetti chiave, dal punto di vista biochimico, della malattia di Tangier è un efflusso di colesterolo difettoso dalla membrana plasmatica delle cellule verso gli accettori extracellulari. La mancata attività di ABCA1 previene la lipidazione dell’apoA-I e, di conseguenza, la formazione di HDL. Nel plasma dei soggetti con la malattia di Tangier sono presenti solo prebeta-HDL mentre le HDL sferiche e mature sono totalmente assenti; gli eterozigoti presentano particelle HDL di piccole dimensioni, povere di colesterolo e relativamente arricchite in apoA-I. I portatori di mutazioni nel gene che codifica per il trasportatore ABCA1 presentano valori di vasodilatazione flussomediata dell’arteria brachiale (FMD) notevolmente ridotti rispetto ai soggetti controllo, indicativi di una compromissione del sistema di regolazione del tono vasale.
Nei soggetti con un solo allele mutato (eterozigoti) la perdita di funzionalità di ABCA1 porta ad un disordine molto più comune e relativamente più lieve, che determina un’ampia varietà di fenotipi clinici e biochimici, in base al tipo di mutazione presente nei portatori.
Mutazioni del gene APOA1
Il gene APOA1 è localizzato sul cromosoma 11 (11q23-q24) e codifica per un polipeptide di 243 aminoacidi. Finora sono state identificate più di 60 mutazioni a carico di questo gene; più della metà di esse sono associate a bassi livelli di HDL-C e principalmente localizzate nella porzione centrale dell’APOA1.
L’apoA-IMilano (apoA-IM), caratterizzata dalla sostituzione dell’arginina in posizione 173 con una cisteina, è stata la prima mutante dell’APOA1 ad essere descritta nell’uomo ed è certamente la variante meglio caratterizzata. Tutti i portatori identificati ad oggi sono eterozigoti per la variante e presentano ridotti livelli plasmatici di HDL-C, un deficit parziale di LCAT e livelli variabili di ipertrigliceridemia. Le HDL dei portatori dj apoA-IM sono piccole, dense e arricchite in particelle discoidali contenenti la forma dimerica della variante. I valori di vasodilatazione sono paragonabili a quelli dei soggetti controllo. Studi in vitro hanno mostrato anche una preservata funzionalità endoteliale nonostante la severa ipoalfalipoproteinemia.
Mutazioni del gene LCAT
Il gene LCAT è localizzato sul cromosoma 16 (16q22), e codifica per una glicoproteina di 416 residui aminoacidici. Mutazioni a carico del gene che codifica per LCAT causano una parziale o completa perdita di attività enzimatica e ipoalfalipoproteinemia. I portatori di due alleli LCAT mutati (omozigoti) presentano una severa riduzione dei livelli plasmatici di HDL-C associata a molteplici alterazioni nella struttura delle HDL e nella distribuzione delle sottoclassi HDL, con una selettiva deplezione delle particelle di grandi dimensioni contenenti apoA-II e la predominanza di piccole prebeta-HDL. I soggetti con un solo allele mutato (eterozigoti) sono caratterizzati da livelli plasmatici di HDL-C mediamente bassi e da un arricchimento in piccole particelle prebeta-HDL.
Mutazioni del gene CETP
Il gene della CETP, localizzato sul cromosoma16 (16q21), codifica per una glicoproteina idrofobica di 476 aminoacidi. Ad oggi, sono state descritte circa 40 mutazioni di questo gene, la maggior parte delle quali identificata in soggetti giapponesi. I portatori di due alleli mutati (omozigoti) presentano livelli plasmatici di HDL-C cinque volte superiori alla norma. La completa assenza di attività enzimatica della CETP impedisce il trasferimento di esteri del colesterolo dalle HDL alle altre lipoproteine determinando così l’accumulo di questi esteri del colesterolo nelle HDL stesse, che divengono, in questo modo, particolarmente grandi. I portatori eterozigoti mostrano un aumento dei livelli plasmatici di HDL-C e di APOA1 e un incremento della dimensione delle HDL.
Effetti delle mutazioni sulle attività del sistema HDL
Gli studi fin qui a disposizione suggeriscono che nei difetti genetici caratterizzati da HDL di piccole dimensioni e arricchite in particelle prebeta il sistema HDL mantiene una corretta funzionalità, mentre nel difetto di CETP in cui si accumulano particelle molto grandi il sistema HDL sembra meno funzionale.
L’attività antiossidante delle HDL isolate da soggetti con mutazioni a carico dei geni che codificano per APOA1, ABCA1 e LCAT è fortemente ridotta rispetto all’attività riscontrata nelle HDL dei soggetti controllo.
L’ispessimento medio-intimale (IMT) carotideo, che rappresenta un valido marcatore di aterosclerosi, è stato misurato in numerosi portatori di disordini genetici di HDL. I portatori dell’apoA-IM presentano valori di IMT carotideo analoghi a quelli di soggetti controllo di pari età e genere. Al contrario, i portatori della variante L202P dell’APOA1 presentano IMT carotideo aumentato e associato a elevato rischio cardiovascolare. Ulteriori studi ancora da approfondire sembrano indicare che le mutazioni nei geni LCAT e CETP non hanno effetto sull’IMT carotideo.