Fattori di rischio dell’aterosclerosi
Quali sono i fattori di rischio dell’aterosclerosi? Approfondiamoli!
Alcuni fattori tendono a raggrupparsi nella sindrome metabolica, che comprende obesità addominale, dislipidemia aterogenica, ipertensione, insulino-resistenza, stato protrombotico e stato proinfiammatorio nei pazienti sedentari. L’insulino-resistenza non è sinonimo di sindrome metabolica, ma può esserne la chiave eziologica.
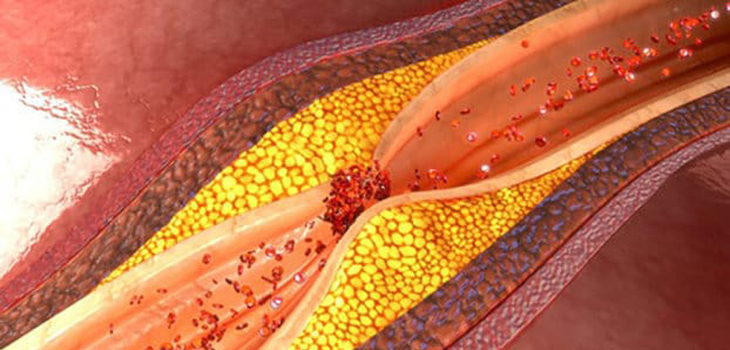
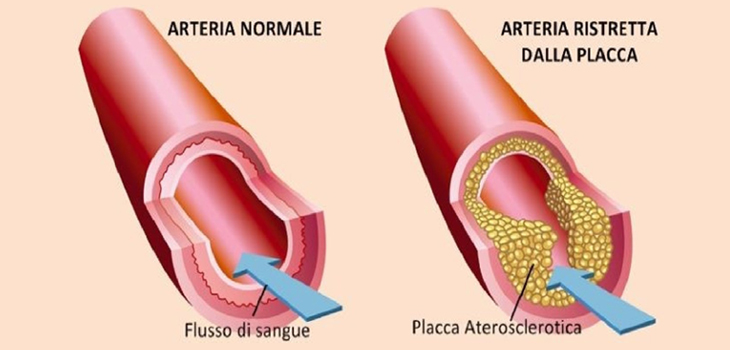
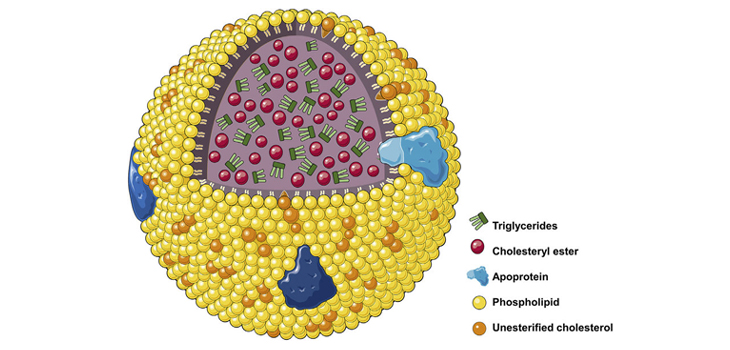
Dislipidemie: livelli elevati di colesterolo totale o di lipoproteine a bassa densità LDL o livelli ridotti di lipoproteine ad alta densità HDL
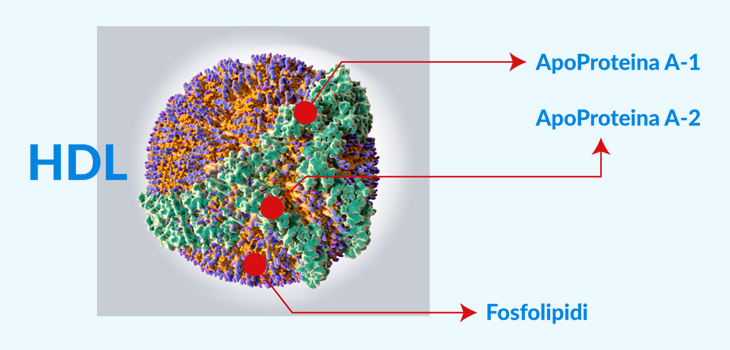
Nelle dislipidemie aumentano i processi di captazione subendoteliale e di ossidazione delle LDL; i lipidi ossidati stimolano la sintesi di molecole di adesione e di citochine infiammatorie e possono avere potere antigenico, stimolando la risposta immunitaria T-mediata e l’infiammazione nella parete arteriosa. Anche se in precedenza si riteneva che le lipoproteine ad alta densità (HDL) proteggessero dall’aterosclerosi attraverso il trasporto inverso di colesterolo e trasportando enzimi antiossidanti, che possono degradare e neutralizzare i lipidi ossidati, recenti evidenze da studi randomizzati e genetici suggeriscono un ruolo molto meno importante per le lipoproteine ad alta densità nell’aterogenesi. Il ruolo dell’ipertrigliceridemia nell’aterogenesi è complesso, sebbene possa avere un piccolo effetto indipendente.
Ipertensione
L’ipertensione può causare infiammazione vascolare tramite meccanismi mediati dall’angiotensina II. L’angiotensina II stimola le cellule endoteliali, le cellule muscolari lisce vascolari e i macrofagi a sintetizzare mediatori proaterogeni, come le citochine proinfiammatorie, gli anioni superossido, fattori protrombotici, fattori di crescita e recettori LDL lectina-simili ossidati.
Diabete
Il diabete porta alla formazione dei prodotti finali della glicazione avanzata, che aumentano la sintesi di citochine proinfiammatorie da parte delle cellule endoteliali. Lo stress ossidativo e i radicali reattivi dell’ossigeno, prodotti nel diabete, danneggiano direttamente l’endotelio e promuovono l’aterogenesi. Un livello elevato delle piccole e dense LDL, caratteristica del diabete, ha un alto potere aterogeno
Fumo di sigaretta
La nicotina e altre sostanze chimiche presenti nel fumo di tabacco hanno un effetto tossico sull’endotelio vascolare. Il fumo, compreso quello passivo, aumentano sia la reattività piastrinica sia i livelli plasmatici di fibrinogeno e dell’ematocrito (aumentata viscosità sanguigna). Il fumo aumenta le LDL e riduce le HDL, stimola la vasocostrizione, particolarmente pericolosa nelle arterie già stenotiche per l’aterosclerosi. Le HDL aumentano di circa 6-8 mg/dL entro 1 mese dall’abolizione del fumo.
Lipoproteina(a)
La lipoproteina(a) o Lp(a) è pro-aterogenica ed è un fattore di rischio indipendente per le malattie cardiovascolari, inclusi l’infarto miocardico, l’ictus e la stenosi valvolare aortica. Ha una struttura simile all’LDL, ma ha anche come componente un’apolipoproteina (a) idrofila legata in modo covalente a un’apolipoproteina idrofobica B100. I livelli di lipoproteina sono geneticamente determinati e rimangono abbastanza stabili per tutta la vita. Livelli di lipoproteina(a) superiori a 50 mg/dL sono considerati patologici.
Apolipoproteina B
L’apolipoproteina B (apoB) è una particella con due isoforme: apoB-100, che è sintetizzata nel fegato, e apoB-46, che viene sintetizzata nell’intestino. L’ApoB-100 è in grado di legare il recettore LDL ed è responsabile del trasporto del colesterolo. È anche responsabile del trasporto di fosfolipidi ossidati e ha proprietà proinfiammatorie. La presenza della particella apoB all’interno della parete arteriosa è ritenuta essere l’evento iniziale per lo sviluppo delle lesioni aterosclerotiche.
Proteina C-reattiva
Livelli elevati di proteina C-reattiva non predicono in modo affidabile l’estensione dell’aterosclerosi, ma possono predire un’aumentata probabilità di eventi ischemici. In assenza di altre malattie infiammatorie, elevati livelli possono indicare un aumentato rischio di rottura, ulcerazione o trombosi della placca aterosclerotica o un’aumentata attività linfocitica e macrofagica.
Infezioni batteriche o virali
Le infezioni da C. pneumoniae, Helicobacter pylori o altre infezioni batteriche o virali possono causare una disfunzione endoteliale attraverso l’infezione diretta, l’esposizione all’endotossina o la stimolazione dell’infiammazione sistemica o subendoteliale.
Malattia renale cronica
La malattia renale cronica promuove lo sviluppo di aterosclerosi attraverso svariati percorsi, tra cui l’ipertensione e il peggioramento dell’insulino-resistenza; diminuiti livelli di apolipoproteina A-I; ed aumentati i livelli di lipoproteina, omocisteina, fibrinogeno e proteina C-reattiva.
Trapianto cardiaco
Un trapianto cardiaco è spesso seguito da un’accelerata aterosclerosi coronarica, che è probabilmente correlata al danno endoteliale immuno-mediato. Un’accelerata aterosclerosi coronarica si osserva anche dopo la radioterapia del torace ed è probabilmente il risultato di danni endoteliali indotti dalle radiazioni.
Iperomocisteinemia
L’iperomocisteinemia per esempio a causa di carenza di folati o di un difetto genetico metabolico hanno un aumentato rischio di aterosclerosi. Tuttavia, non si ritiene che l’iperomocisteinemia stessa causi l’aterosclerosi e la causa dell’associazione tra elevati livelli di omocisteina e aterosclerosi non è chiara.